- Fast Facts
- First Description
- Who gets Henoch-Schönlein Purpura (the “typical” patients)?
- What causes Henoch-Schönlein Purpura?
- How is Henoch-Schönlein Purpura diagnosed?
- Treatment and Course of Henoch-Schönlein Purpura
- Living with Henoch-Schönlein Purpura
Fast Facts
- HSP is usually self-limited. Therefore, treatment is not indicated in all cases, and full recovery is the rule.
- HSP is more common in children than adults, but has a tendency to be more severe when it occurs in adults.
- In a small minority of cases, HSP can cause severe kidney or bowel disease.
First Description
Dr. William Heberden, a London physician, described the first cases of Henoch-Schönlein purpura (HSP) in 1801. In describing HSP, Heberden wrote of a 5-year old boy who “…was seized with pains and swellings in various parts…He sometimes had pains in his belly with vomiting…and the urine was tinged with blood. Presently, the skin of his leg was all over full of bloody points” (purpura). The young boy suffered all four disease hallmarks of HSP: arthritis, gastrointestinal involvement, kidney inflammation, and purpura. Johann Schönlein (1837) and Edouard Henoch (1874) reported additional cases decades after Heberden. They recognized that the disorder often followed upper respiratory tract infections and was not always self-limited, sometimes progressing to serious kidney involvement.
Who gets Henoch-Schönlein Purpura (the “typical” patient)?
Usually, HSP affects a child shortly after an upper respiratory infection has resolved.
HSP is the most common form of vasculitis in children, with an annual incidence on the order of 140 cases/million persons. The mean age of patients with HSP is 5.9 years.
What causes Henoch-Schönlein Purpura?
In two-thirds of the cases, the disease follows an upper respiratory tract infection, with onset an average of ten days after the start of respiratory symptoms. Despite this association, no single microorganism or environmental exposure has been confirmed as an important cause of HSP.
How is Henoch-Schönlein Purpura Diagnosed?
Purpura not due to a low platelet count, caused by inflammation in blood vessels of the skin, is the hallmark of HSP. The tetrad of purpura, arthritis, kidney inflammation, and abdominal pain is often observed. However, all four elements of this tetrad are not required for diagnosis. The microscopic hallmark of HSP is the deposition of IgA (an antibody found in blood, saliva, tears, etc.) in the walls of involved blood vessels.
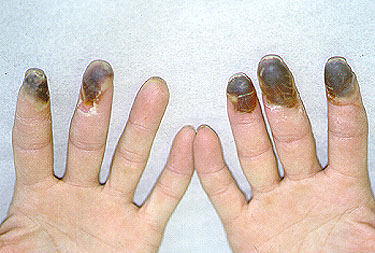
More than 90% of cases occur in children. The disease usually resolves within a few weeks. However, adult cases are sometimes more difficult. Skin manifestations are more variable in adults, and sometimes symptoms in adults endure longer [Figure 1, 2].

Figure 1. Pustular lesions. These can occur in HSP, but they are more common with the adult form of HSP.

Figure 2. Vesiculobullous lesions These are also more common with the adult form of HSP.
Adults are more prone to permanent kidney damage. However, patients can take some comfort in knowing that fewer than 5% of patients with HSP develop progressive renal insufficiency.
HSP can be mimicked by other forms of systemic vasculitis that are more often life-threatening. Granulomatosis with polyangiitis and microscopic polyangiitis can also present with purpura, arthritis, and renal inflammation. These disorders both have the potential for serious involvement of other organs (for example, the lungs, eyes, and peripheral nerves) and carry more dire renal prognoses. Therefore, it is very important to distinguish the difference by performing a careful evaluation including bloodwork, urinalysis, chest imaging, and possibly biopsies. HSP may be misdiagnosed as another form of vasculitis – most commonly hypersensitivity vasculitis – because of the frequent failure to perform direct immunofluorescence (DIF) testing on skin biopsy and the consequent failure to detect IgA.
Treatment and Course of Henoch-Schönlein Purpura
NSAIDs may alleviate arthralgias but can aggravate gastrointestinal symptoms, and should be avoided in any patient with renal disease. Dapsone (100 mg/day) may be effective in cases of HSP, perhaps through disrupting the abnormal immune response. Although steroids have not been evaluated rigorously in HSP, they appear to ease joint and gastrointestinal symptoms, in many (but not all) patients. Steroids, however, do not appear to improve the rash; although usually, over weeks to months, the recurrent bouts of purpura usually resolve on their own.
Living With HSP
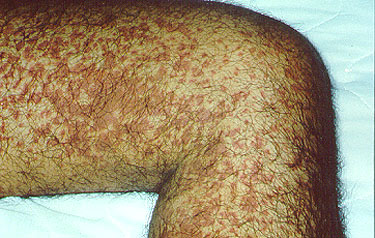
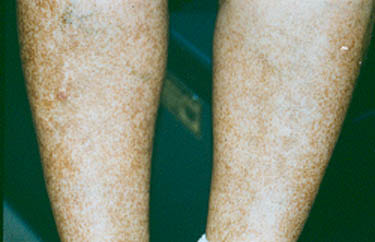
Supportive care may involve a short course of prednisone or an NSAID, such as naprosyn or ibuprofen, if the kidneys are not involved. Keeping the legs elevated may help prevent purpura during flares of active disease. Additionally, many patients’ purpura will recur after they start to feel better and become more active, inherently increasing their exposures to very minor trauma (e.g. jogging, leg shaving, increasing gravity exposures). Often, the recurring purpura is less prevalent [Figure 3], and additional HSP symptoms are often absent. In many fewer cases, primarily in adults, HSP can progress from hematuria (blood in the urine) to renal insufficiency (decreased kidney function). HSP patients who experience this symptom should be followed more closely, with regular testing of their urine for blood and protein. Recurrences, found in 33% of patients, usually develop within the first few months after resolution of the first bout.

Figure 3. Palpable purpura . Occurring in a more diffuse pattern.

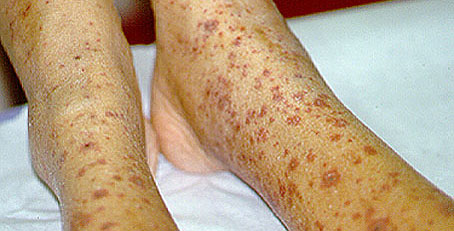
Figure 4. Palpable purpura . Here they are occurring in a very dense pattern with coalescing lesions.

Figure 5. Swelling around the hand and wrist . Although arthralgias are more common in HSP, arthritis can occur as well as periarticular swelling, such as the tenosynovitis shown here.

Figure 6. Swelling around the ankle and foot .

Figure 7. Palpable purpura can appear in many different patterns . This picture shows a denser distribution with a sharp demarcation caused by what is known as Koebner’s Phenomenon (Minor trauma, such as the elastic band in one’s sock, can cause such a pattern). In this case, the “trauma” was caused by the patient’s shaving of her legs, leading to the eruption of purpura in the area of skin where the razor had passed. Sufficient pressure, such as this, causes the rupture of inflamed blood vessels.

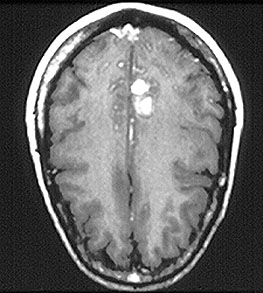
Figure 8. CT of abdomen showing bowel edema . This image is of a distended large bowel. The characteristic dips between haustra (bowel sections) are less pronounced because of the swelling / inflammation seen in HSP.

Figure 9. Formally known as DIF (Direct Immunofluorescence) testing. This picture shows immunofluorescence testing of a skin biopsy, IgA positive. Palpable purpura should be biopsied, and two fresh samples should always be sent for testing (an adequate biopsy should be large enough to divide; one for H&E (hematoxylin and eosin) staining, and one for DIF testing.

Figure 10. Arm rash . It is more common to have a purpuric outbreak on the lower extremities. However, an outbreak can occur on the abdomen, chest, or as in the case with this woman, on the upper extremities. Note the hive-like lesions that appear larger than the papules. The rash may also be itchy.

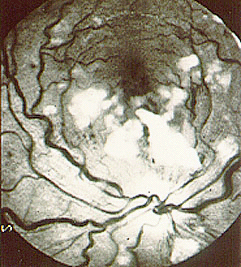
Figure 11. Colonoscopy of HSP-affected bowel . This image shows what the lining of the bowel could look like when it is inflamed and swollen, as in HSP. Looks painful…it is.