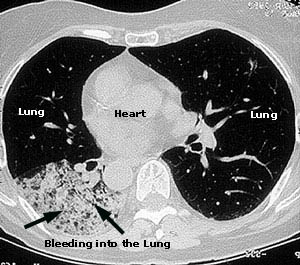
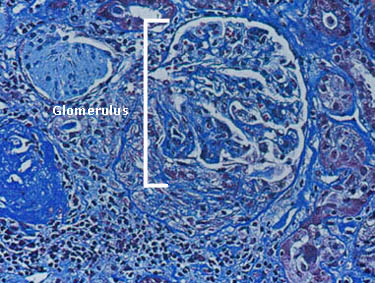
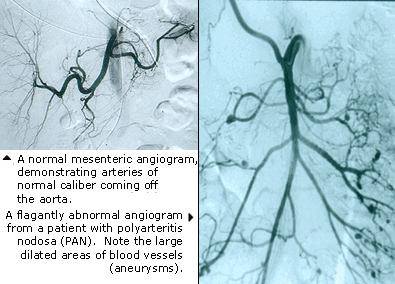
What is IVIG/SCIG?
Intravenous immunoglobulin (IVIG) is a therapy consisting of pooled antibodies (immunoglobulin) obtained from healthy donors that is given as an infusion by vein. This same therapy can also be given as a subcutaneous injection (SCIG) rather than an intravenous one.
How does it work?
IVIG and SCIG are often used to treat patients with immunodeficiency syndromes, which are genetic or acquired conditions that lead to low immunoglobulin levels. For these patients, IVIG/SCIG provide the protective effect of antibodies that they otherwise lack.
In treating vasculitis, we sometimes encounter the need for IVIG/SCIG due to the use of Rituximab – a drug that targets B cells. In some patients, the long-term use of rituximab may lead to an acquired deficiency of immunoglobulins. By combining IVIG/SCIG with rituximab, we are able to continue to provide patients with the immunosuppressive benefit of rituximab, while compensating for the increased risk of infection by giving IVIG/SCIG.
How is IVIG/SCIG given?
IVIG is often given as a home infusion. SCIG is given as a subcutaneous injection. These treatments are generally given once per month.
Side effects:
These treatments carry a risk of blood clot, renal injury, and headaches. IVIG constitutes a large fluid challenge, and therefore may not be appropriate for patients with heart or kidney failure.