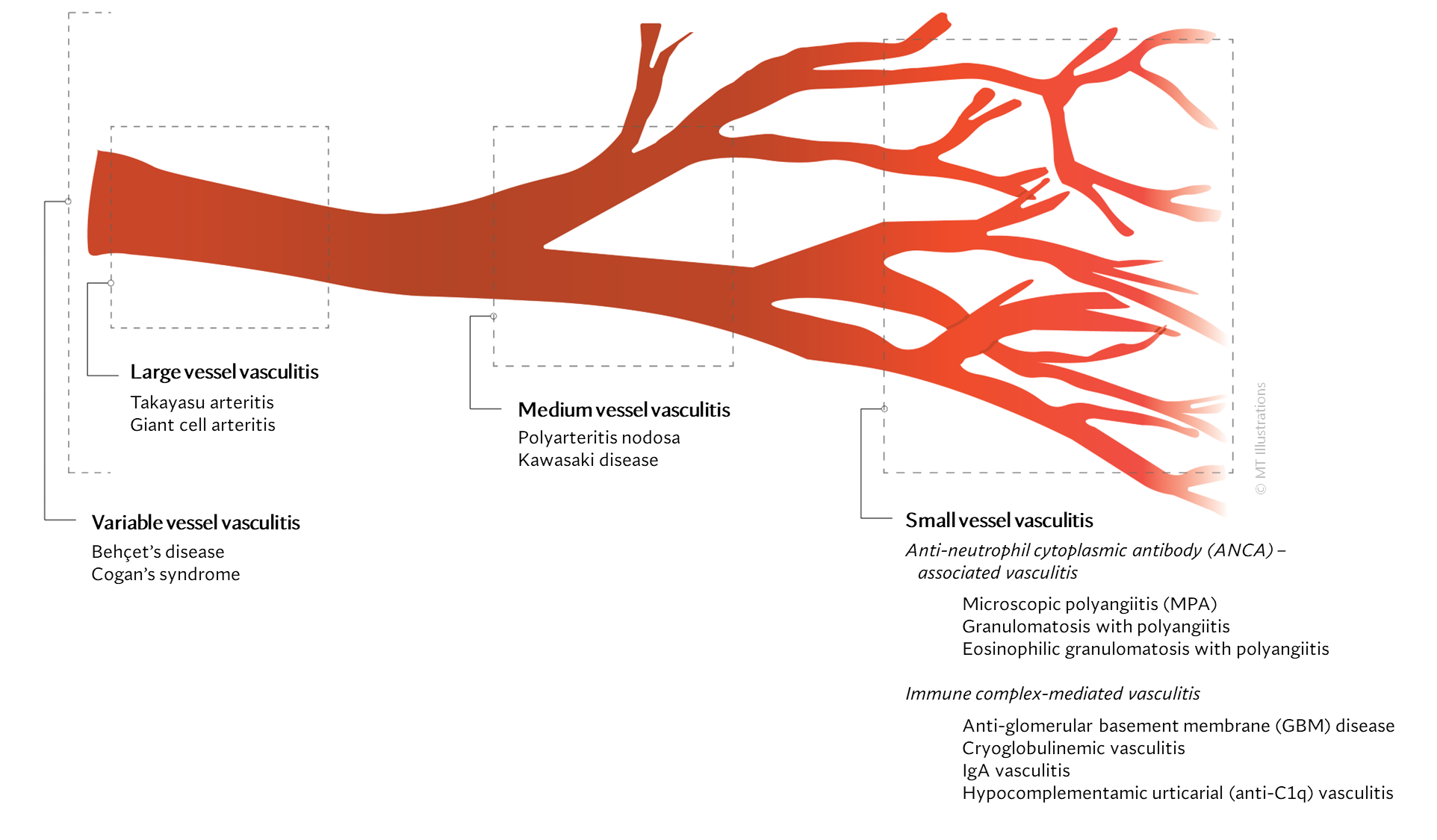
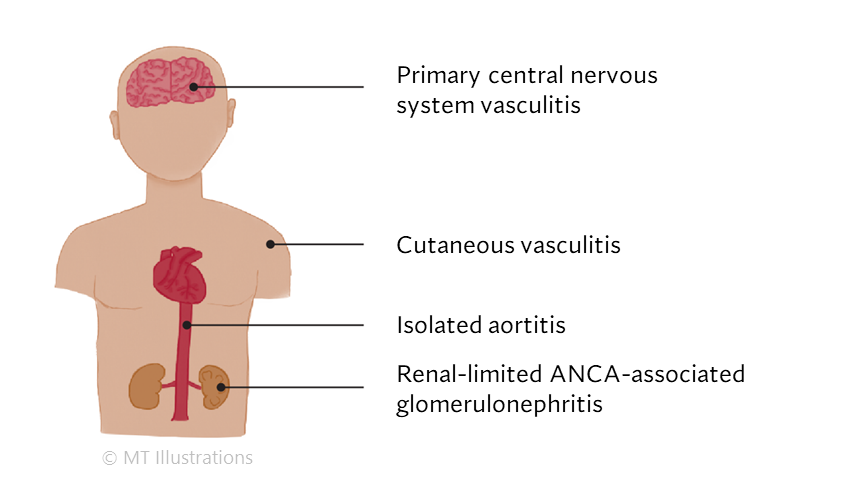
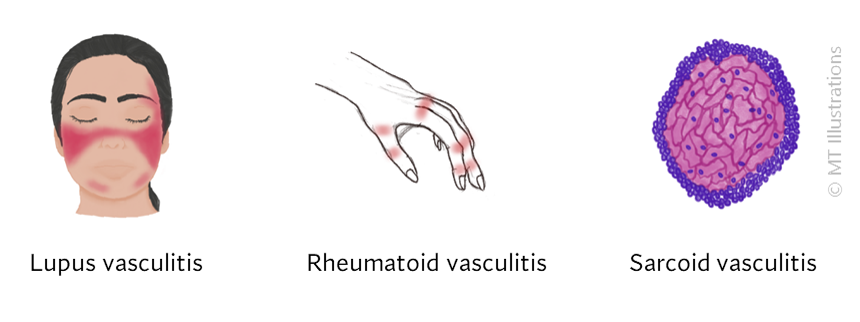
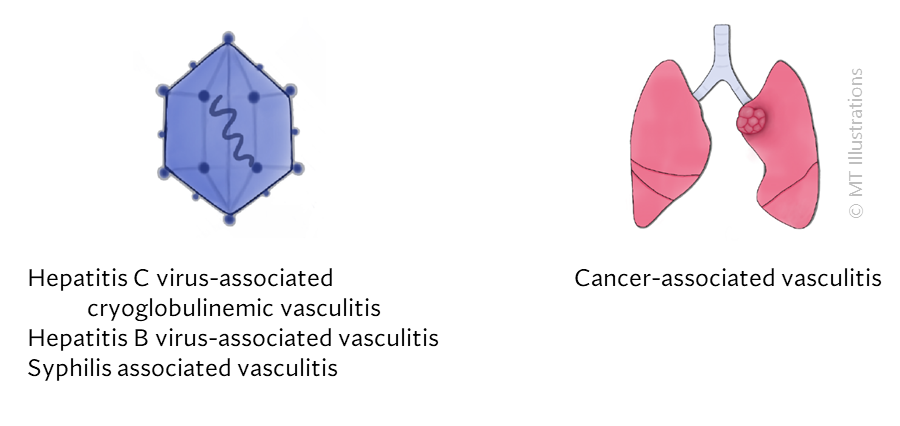
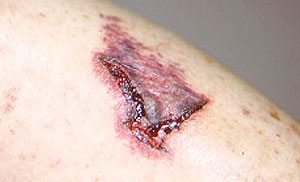
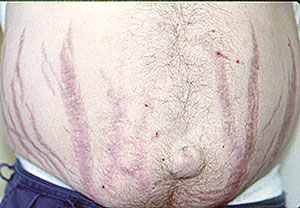
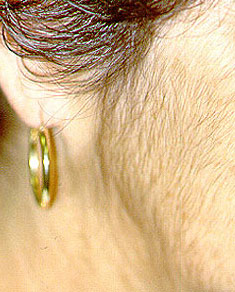
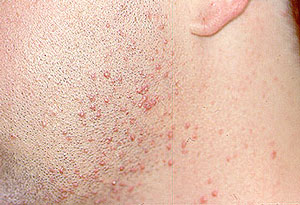
Our center is composed of dedicated physicians, research coordinators and patient care coordinators who serve patients with vasculitis. Many patients who receive these diagnoses have never previously heard the term “vasculitis” or met other patients with the same condition. The vasculitis syndromes (known together as “the vasculitides”) are a group of diseases that can affect every organ system, and occur in people of all ages, genders and backgrounds. Because these diseases are relatively rare and can present in many different ways, the diagnosis of vasculitis is often difficult to reach, and many patients suffer a period of uncertainty prior to finally arriving at a diagnosis of vasculitis. In our mission of serving all patients with vasculitis, we consider the provision of clear and accurate information to be one of our most important responsibilities.
At this Website you will find:
- explanations of vasculitis in lay terms
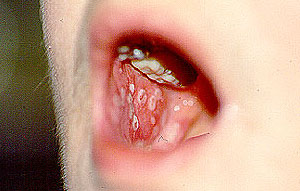
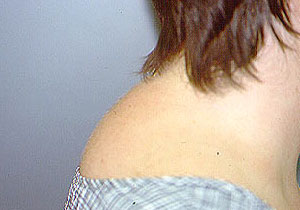
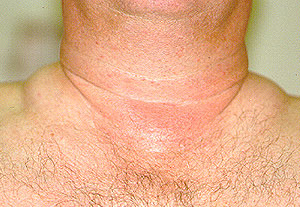
- specific discussions of individual diseases
- a review of the common therapies for vasculitis
- answers to commonly asked questions
- information on how to make an appointment to be seen in the Johns Hopkins Vasculitis Center
- information about ongoing research at the Johns Hopkins Vasculitis Center
- ways in which you can contribute to advancing research and progress in vasculitis.
Please note that this Website is sponsored through the generosity of various friends of the Johns Hopkins Vasculitis Center. We update the Vasculitis Center Website regularly and strive to provide solid, usable information on various types of vasculitis, treatments, and support resources.
Thank you for visiting our Website. We hope you will find it accessible and useful as you learn about these challenging diseases.
Yours truly,
Brendan Antiochos, MD
Assistant Professor of Medicine
Johns Hopkins University School of Medicine,
Division of Rheumatology
Director, The Johns Hopkins Vasculitis Center