- What causes vasculitis?
- What is going to happen to me?
- Is vasculitis curable?
- Is vasculitis hereditary?
- Does diet affect vasculitis?
- Will my vasculitis return?
- How should I guard against the occurrence of a disease flare?
- Why do I have to have bloodwork checked frequently?
What causes vasculitis?
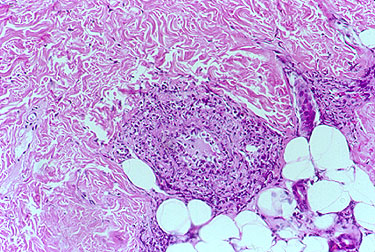
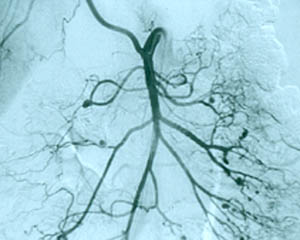
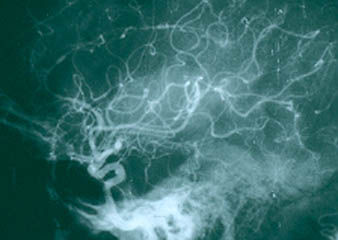
The causes of most forms of vasculitis remain unknown. Infections are strongly suspected of playing a role in in forms such as the association of hepatitis B (a virus) and polyarteritis nodosa, and hepatitis C (another virus) and cryoglobulinemic vasculitis. Bacterial infections have been suspected of playing a possible role in granulomatosis with polyangiitis (GPA, formerly known as Wegener’s) which is the reason that some patients with GPA that is limited to the upper respiratory tract are treated only with an antibiotic, Bactrim (trimethoprim/sulfamethoxazole). A general theory that applies to many types of vasculitis is that the disease results from the occurrence of a particular infection in a person whose genes (and other factors) make him/her susceptible to developing vasculitis.
What is going to happen to me?
The course of vasculitis is often difficult to predict. Some types of vasculitis may occur only once and do not return. Other types are prone to recurrences. For all patients with vasculitis, it is essential to be evaluated by physicians who are experienced in the treatment of these diseases. Vasculitis is treatable, and many patients achieve remissions through treatment. It is important to balance the types of medications necessary to control the disease and the risk of side effects that those medicines often bring. A primary aim of several ongoing new studies in vasculitis is to find drugs that help maintain remission.
Is vasculitis curable?
Most forms of vasculitis are treatable if detected early enough, before substantial organ damage has occurred. While often effective, however, the treatments remain imperfect and require improvement. Further research is needed in all forms of vasculitis. Greater knowledge of these diseases will lead to better treatments and, some day, to cures.
Will my children or other family members get it?
Vasculitis is not contagious. One cannot acquire vasculitis from contact with a vasculitis patient. In addition, despite the fact that genes probably play a role in susceptibility to some forms of vasculitis, it is unusual for vasculitis to occur in more than one member of the same family. Thus, vasculitis is not a heritable disorder. All of these points illustrate the fact that the causes of vasculitis are complex. In all likelihood, patients develop vasculitis because of the simultaneous occurrence of multiple risk factors, most of which remain poorly understood.
Does diet affect vasculitis?
This is one of the most commonly-asked questions by patients with vasculitis. All patients want to do whatever is within their power to help treat their disease. Unfortunately, there is presently no evidence that a person’s diet affects susceptibility to vasculitis, or that consuming or avoiding certain foods or beverages affects the course of the disease. In general, we advocate eating a balanced healthy diet rich in protein and vegetables. Avoidance of excessive empty calories, processed foods, and sugars may be very important, particularly in patients on steroids who are at risk for weight gain.
Will my vasculitis return?
After patients achieve remission from their vasculitis, it is logical for them to wonder if their disease will ever return. The answer, which is often difficult to give with certainty, depends in large part on the patient’s specific type of vasculitis. For example, some types of vasculitis, such as Henoch-Schönlein purpura (HSP) or vasculitis caused by a medication, are often self-limited and resolve on their own. Other forms of vasculitis (e.g., Buerger’s disease, a disease strongly associated with cigarette smoking) resolve with institution of the definitive treatment: smoking cessation.
However, other forms of vasculitis behave less predictably and never come back in some patients but recur frequently in others. Granulomatosis with polyangiitis (GPA), giant cell arteritis (GCA), Takayasu arteritis, microscopic polyangiitis, and many other types of vasculitis fall into the category of diseases that have periods of quiescence and periods of flare. Disease flares in vasculitis can be mild (rash, minor joint pains) or severe (renal failure, skin ulcers). Flares may occur if medications are discontinued or dosage is lowered. Flare may occur in the context of infection. Often the reason for disease flare is unknown.
At the present time, the ability of doctors to predict who will suffer disease flares and who will maintain in long-term remissions (or be cured) needs refinement. Progress in this area will come through research.
How should I guard against the occurrence of a disease flare?
We believe that several points are worth keeping in mind:
First, the symptoms of flares are usually very similar those experienced at the onset of disease. If headaches signaled the beginning of giant cell arteritis, then the recurrence of headaches may indicate a disease flare. If leg ulcers began as painful red lumps on the leg the first time, then the return of painful red lumps may mean that vasculitis is back. Patients must become experts about their own manifestations of vasculitis so that they can recognize them immediately, consult their doctors, and begin appropriate treatment before serious damage occurs.
Second, we believe that patients truly know and understand their own bodies. It is important to discuss new or changing symptoms with your physicians. Together, patients and physicians can determine if new symptoms truly represent a vasculitis flare or if the cause is something equally as likely (medication side effect, infection, or other common medical issues).
Finally, because vasculitis treatments require careful monitoring by doctors, patients should discuss any changes in treatment with their physicians. Increasing or decreasing medications without consulting a physician may lead to trouble.
Why do I have to have bloodwork checked frequently?
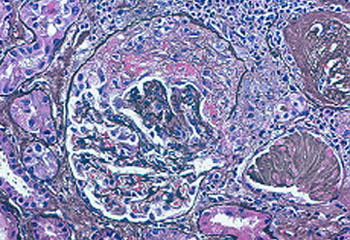
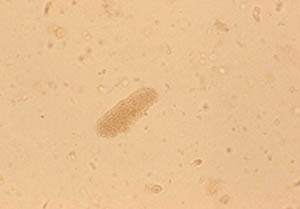
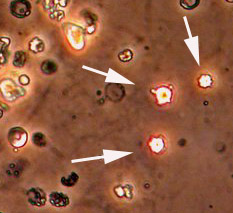
Blood tests are helpful to monitor for the return of vasculitis by keeping a watchful eye on important parameters such as kidney function, liver tests, and markers of inflammation (ESR and CRP). Blood tests are also very important to ensure that medications are not causing any side effects such as liver irritation or low blood counts.
How often should my blood be checked?
This depends on the specific medicine or medicines that you take. Patients on cyclophosphamide (Cytoxan) should have their counts checked every week. Patients on most other kinds of medications used to treat vasculitis (Methotrexate, Azathioprine) usually only need to have their blood work checked monthly. If some laboratory tests are abnormal or nearly so, then more frequent monitoring may be required.
What type of tests do we check?
Regardless of the type of vasculitis and the exact type of medication that a patient takes, similar types of tests are monitored. These tests are:
- a complete blood count;
- tests of kidney function including a urinalysis; and
- liver function tests.
The table below outlines the importance behind checking each of these tests.
| Type of Test | What should be checked | Why? |
| Complete Blood Count (“CBC”) |
|
|
| Kidney Function |
|
|
| Urinalysis |
|
|
| Liver Function |
|
|