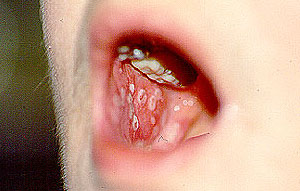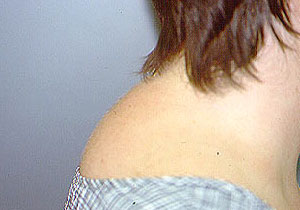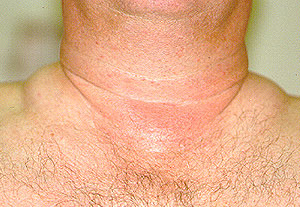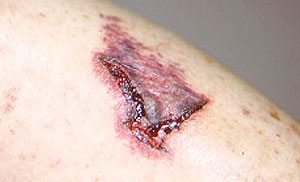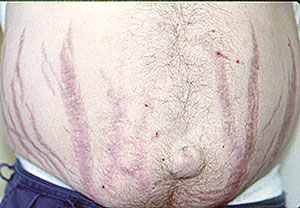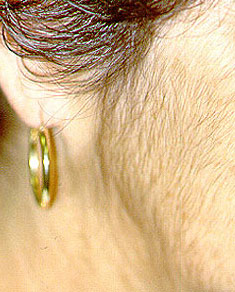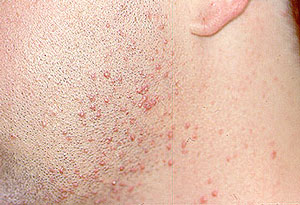
Dear Vasculitis Center Website Visitor:

Welcome to the Johns Hopkins Vasculitis Center Website. This Website, maintained by the Physicians, Research Coordinators, and Patient Care Coordinators at our Center, is designed to provide information for patients with vasculitis in language that non-medical people can understand. We recognize that many patients with vasculitis have never heard of their disease before they became sick and that, owing to the relative rarity of some types of vasculitis, most physicians have little experience treating the disorders. Few support groups for vasculitis patients exist, and there is a shortage of literature about these diseases written for lay people. Consequently, most patients find reliable information about vasculitis difficult to come by.
At this Website you will find:
- explanations of vasculitis in lay terms
- specific discussions of individual diseases
- a review of the common therapies for vasculitis
- answers to commonly asked questions
- information on how to make an appointment to be seen in the Johns Hopkins Vasculitis Center
- information about ongoing research at the Johns Hopkins Vasculitis Center
- ways in which you can contribute to advancing research and progress in vasculitis.
Please note that this Website is sponsored through the generosity of various friends of the Johns Hopkins Vasculitis Center. We update the Vasculitis Center Website regularly and strive to provide solid, usable information on various types of vasculitis, treatments, and support resources.
Thank you for visiting our Website. We hope you will find it accessible and useful as you learn about these challenging diseases.
Yours truly,
Philip Seo, MD, MHS
Assistant Professor of Medicine
Johns Hopkins University School of Medicine,
Division of Rheumatology
Director, The Johns Hopkins Vasculitis Center