- First Description
- Who gets Polyarteritis Nodosa (the “typical” patients)?
- Classic symptoms of Polyarteritis Nodosa
- What causes Polyarteritis Nodosa?
- How is Polyarteritis Nodosa diagnosed?
- Treatment and Course of Polyarteritis Nodosa
- In medical terms, by David Hellmann, M.D.
First Description
The first description of this disease dates back to 1866 when Kussmaul and Maier identified a condition that consisted of “focal, inflammatory, arterial nodules”. They termed this disorder “periarteritis nodosa” because of the inflammation they observed around the blood vessel wall. The name was changed to polyarteritis nodosa (PAN) to underscore the fact that inflammation throughout the entire arterial wall – not just around the wall – is a major disease feature. Polyarteritis nodosa is sometimes termed “systemic necrotizing vasculitis”, but this term is non-specific as other forms of vasculitis also have systemic and necrotizing features.
Who gets Polyarteritis Nodosa (the “typical” patient)?
Most cases of PAN occur in the 4th or 5th decade, although it can occur at any age. Men are twice as likely to be affected than women. A minority of patients with PAN have an active hepatitis B infection. In the rest of the cases, the cause(s) is presently unknown, and the disease is said to be “idiopathic” in nature.
Classic symptoms and signs of Polyarteritis Nodosa
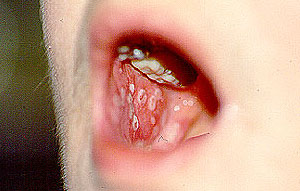
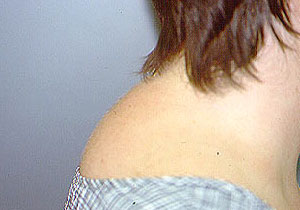
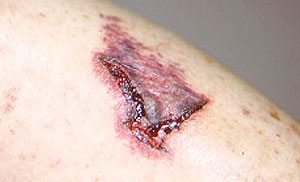
PAN is a multisystem disease that may present with fever, sweats, weight loss, and severe muscle and joint aches/pains. PAN may develop in a subacute fashion, over several weeks or months. Patients may have nonspecific complaints such as fever, malaise, weight loss, anorexia, and abdominal pain. The disease can affect nearly any site in the body, but it has a predisposition for organs such as the skin, kidney, nerves, and gastrointestinal tract. Many patients with PAN have high blood pressure and elevated erythrocyte sedimentation rates (ESR). The presentation of PAN may also include skin abnormalities (rash, ulcers) and peripheral neuropathy (pain, the sensations of burning, tingling, or numbness, or weakness in a hand or foot). However, the disease has a predilection for certain organs and tissues; these are described below.
Nerve
- Peripheral neuropathies are very common (50 to 70%). This includes tingling, numbness and/or pain in the hands, arms, feet, and legs.
- Central nervous system (CNS) lesions may occur 2 to 3 years after the onset of PAN and may lead to cognitive dysfunction, decreased alertness, seizures and neurologic deficits.

Skin
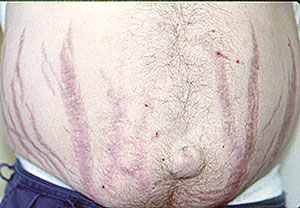
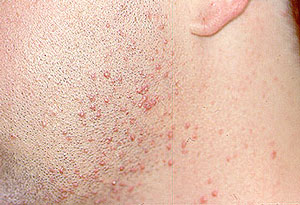
- Skin abnormalities are very common in PAN and may include purpura, livedo reticularis, ulcers, nodules or gangrene.
- Skin involvement occurs most often on the legs and is very painful.

Kidney
- Renal artery vasculitis may lead to protein in the urine, impaired kidney function, and hypertension.
- Small percentage of patients go on to require dialysis.
Gastrointestinal Tract
- Abdominal pain, gastrointestinal bleeding (occasionally is mistaken for inflammatory bowel disease)
- Hemorrhage, bowel infarction, and perforation are rare, but very serious
Heart
- Clinical involvement of the heart does not usually cause symptoms.
- However, some patients develop myocardial infarctions (heart attacks) or congestive heart failure.
Eye
- Scleritis or inflammation in the sclera (white part of the eye)
Genitals
- Testicular infarction
What causes Polyarteritis Nodosa?
Hepatitis B causes a minority of cases of PAN. With the availability of hepatitis B vaccine now, cases of PAN caused by hepatitis B are now rare in the developed world. It is possible that other infections contribute to other cases of PAN, but links between other infections and this disease remain conjectural at the present time.
How is Polyarteritis Nodosa Diagnosed?
Routine laboratory tests may provide important clues to PAN, but there is no single blood test that is diagnostic of this disease. Most patients with PAN have elevated ESRs. Proteinuria (protein in the urine) is common among those with kidney involvement.
If there is skin or muscle/nerve involvement, a skin or muscle/nerve biopsy can be extremely helpful in coming to a definite diagnosis of PAN. Nerve conduction studies are a non-invasive way of identifying nerves that are involved by the inflammation. (These nerves can then be biopsied to confirm the diagnosis). The diagnosis is confirmed by a biopsy showing pathologic changes in medium-sized arteries. The biopsy site may vary. Most biopsies are taken from skin, symptomatic nerve, or muscle. An angiogram of the abdominal blood vessels may also be very helpful in diagnosing PAN. Aneurysms most often affect the arteries leading to the kidneys, liver or gastrointestinal tract.
The American College of Rheumatology (ACR) has established criteria that should be fulfilled if a patient is to be included in a research study of PAN. The criteria are designed to differentiate PAN from other forms of vasculitis. Not all patients have all criterion. Some, in fact, may have only 2 or 3 criteria, yet their physicians are still comfortable classifying their disease as PAN. A committee of ACR physicians selected 10 disease features (criteria) as being those that best distinguish PAN from other vasculitides. In order to be classified as a PAN patient – for the purpose of research studies – a patient should have at least 3 of the 10 ACR criteria.
The American College of Rheumatology 1990 criteria for the classification of Polyarteritis Nodosa
- Weight loss of > 4 kg since beginning of illness
- Livedo reticularis
- Testicular pain or tenderness
- Myalgias, weakness, or leg tenderness
- Mononeuropathy or polyneuropathy
- Development of hypertension
- Elevated BUN or creatinine unrelated to dehydration or obstruction
- Presence of hepatitis B surface antigen or antibody in serum
- Arteriogram demonstrating aneurysms or occlusions of the visceral arteries
- Biopsy of small or medium-sized artery containing granulocytes
Treatment and Course of Polyarteritis Nodosa
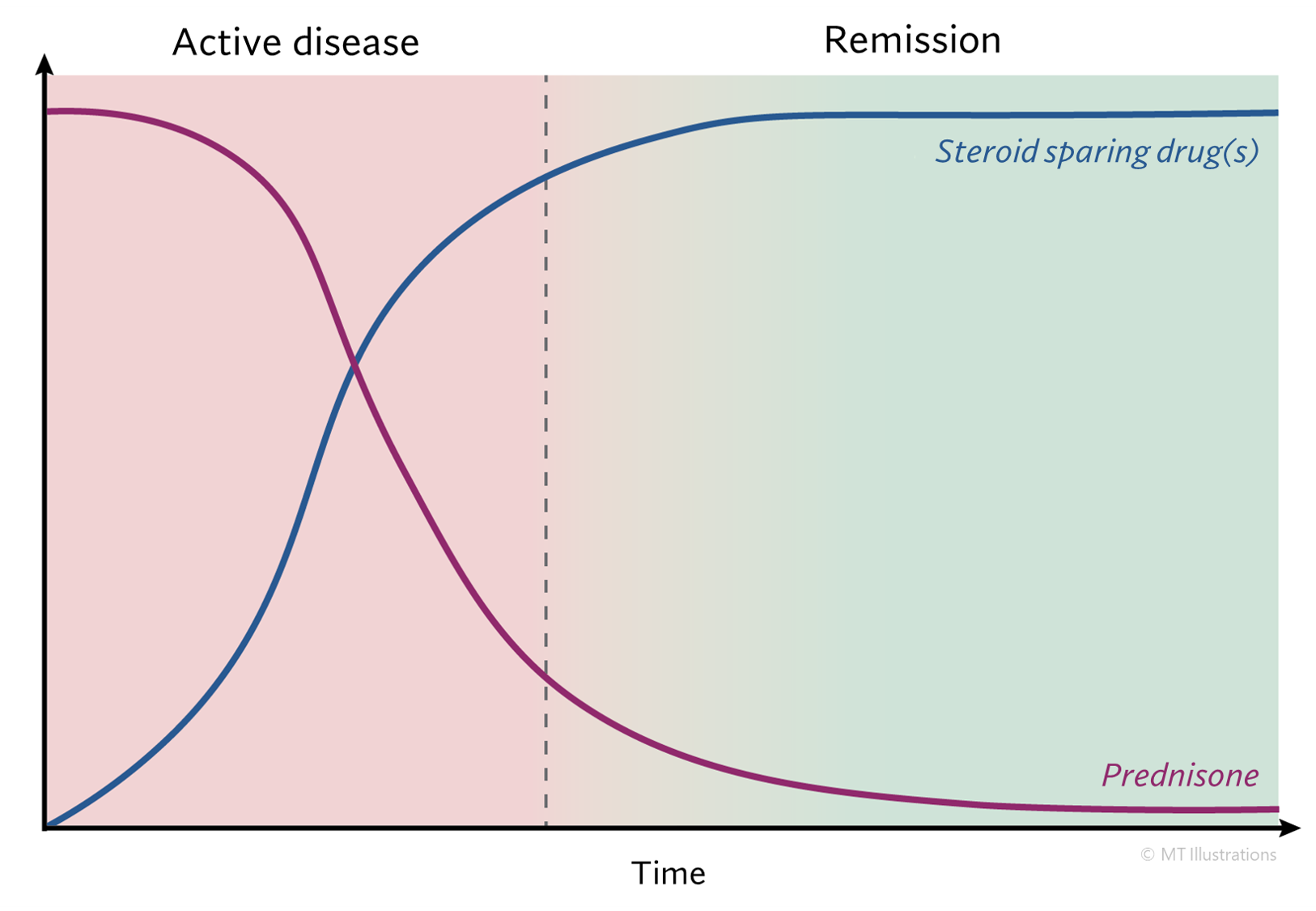
Treatment of PAN has improved dramatically in the past couple of decades. Before the availability of effective therapy, untreated PAN was usually fatal within weeks to months. Most deaths occurred as a result of kidney failure, heart or gastrointestinal complications. However, effective treatment is now available for PAN. After diagnosis, patients are treated with high doses of corticosteroids. Other immunosuppressive drugs are also added for patients who are especially ill. In most cases of PAN now, if diagnosed early enough the disease can be controlled, and often cured.
In medical terms, by David Hellmann, M.D.
A discussion of Polyarteritis Nodosa written in medical terms by David Hellmann, M.D. (F.A.C.P.), for the Rheumatology Section of the Medical Knowledge Self–Assessment Program published and copyrighted by the American College of Physicians (Edition 11, 1998). The American College of Physicians has given us permission to make this information available to patients contacting our Website.
Polyarteritis nodosa is a small– and medium–sized arteritis affecting multiple organs, especially the skin, peripheral nerve, gut, kidney, and heart. The age of onset ranges from childhood to late adulthood but averages 40 years. Polyarteritis nodosa has been associated with active hepatitis B, hepatitis C, or both; therefore, the disease is more common in injection drug users.
Polyarteritis nodosa is probably mediated by deposition of immune complexes. Evidence includes the observation that patients with polyarteritis nodosa associated with hepatitis B or hepatitis C have immune complexes consisting of immunoglobulin and viral antigens circulating in the blood and deposited in inflamed vessels. Moreover, antiviral therapy can remit the vasculitis in some of these patients.
The onset is gradual over weeks to months, and the initial symptoms are often nonspecific. The earliest clues that the patient has vasculitis come usually from the skin (where vasculitis may appear as palpable purpura, livedo reticularis, digital gangrene, or tender nodules), or the peripheral nervous system (where infarction of one mixed motor and sensory nerve after another results in mononeuritis multiplex, one of the most specific clues that a patient has vasculitis). Renal involvement eventually develops in most and is accompanied by hypertension in half of patients, whereas Granulomatosis with Polyangiitis
rarely elevates the blood pressure. Polyarteritis nodosa also commonly involves the gut (abdominal angina, hemorrhage, perforation), heart (myocarditis, myocardial infarction), or eye (scleritis). Rupture of renal or mesenteric micoaneurysms can simulate an acute abdomen.
Confirming the diagnosis requires either biopsy specimen showing small– or medium–sized arteries, or mesenteric arteriography showing microaneurysms or alternating areas of stenosis and dilation. Biopsy of a symptomatic nerve or a symptomatic muscle is 65% sensitive, whereas biopsy of an asymptomatic site is less than 30% sensitive. Because mesenteric angiography is 60% sensitive, it should be done when there is not a symptomatic site to biopsy. Renal biopsy should be avoided unless angiography rules out microaneurysms susceptible to rupture.
Without treatment, almost all affected patients die within 2 to 5 years. Treatment with prednisone (starting at 1 mg/kg daily) and cyclophosphamide (2 mg/kg daily) appeared to revolutionize the outcome of polyarteritis nodosa by achieving 70% 10–year survivals and established this combination of agents as the standard therapy. However, newer studies suggest that prednisone alone may achieve the same high survival as prednisone and cyclophosphamide, although flares were less frequent in patients taking cyclophosphamide. Other studies indicate that the traditional therapy with prednisone and cyclophosphamide should be abandoned in patients with polyarteritis nodosa associated with hepatitis B. Patients treated with the traditional combination respond, but almost all survivors become chronic carriers of hepatitis B and may die later of cirrhosis or variceal bleeding. The newly propsed regimen consists of 2 weeks of prednisone to control the vasculitis, followed by plasmapheresis to remove immune complexes, and accompanied by antiviral therapy with lamivudine to rid the patient of the hepatitis B infection. The long–term value of anti–viral therapy for polyarteritis nodosa associated with hepatitis C is not established.