- First Description
- Who gets Takayasu’s Arteritis (the “typical” patients)?
- Classic symptoms of Takayasu’s Arteritis
- What causes Takayasu’s Arteritis?
- How is Takayasu’s Arteritis diagnosed?
- Treatment and Course of Takayasu’s Arteritis
- What’s new in Takayasu’s Arteritis?
- In medical terms, by David Hellmann, M.D.
First Description
The first case of Takayasu’s arteritis was described in 1908 by Dr. Mikito Takayasu at the Annual Meeting of the Japan Ophthalmology Society. Dr. Takayasu described a peculiar “wreathlike” appearance of blood vessels in the back of the eye (retina). Two Japanese colleagues at the same meeting reported similar eye findings in patients whose wrist pulses were absent. It is now known that the blood vessel malformations that occur in the retina are a response (new blood vessel growth) to arterial narrowings in the neck, and that the absence of pulses noted in some patients occur because of narrowings of blood vessels to the arms. The eye findings described by Dr. Takayasu are rarely seen in patients from North America.
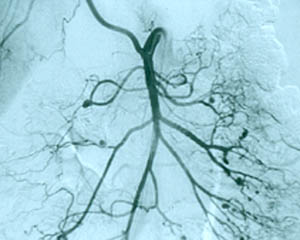
Pictured below is a close–up view of an angiogram of the left vertebral and subclavian arteries in a patient with Takayasu’s arteritis. Note the narrowing and irregularities that occur at several sites, and the “corkscrew” configuration of one vessel segment near the junction of the two arteries. These changes, caused by inflammation in the blood vessel wall, sometimes cause complete blockage of the artery.

Takayasu’s arteritis is occasionally called “pulseless disease”, because of the difficulty in detecting peripheral pulses that sometimes occurs as a result of the vascular narrowings.
Who gets Takayasu’s Arteritis?
The “typical” patient with Takayasu’s arteritis is a woman under the age of 40. There is a 9:1 female predominance in this disease. Although the disease has a worldwide distribution, it appears to occur more often in Asian women.
Takayasu’s arteritis is a rare disease. The best estimates of the disease frequency suggest that 2 or 3 cases occur each year per million people in a population.
Classic Symptoms of Takayasu’s Arteritis
Takayasu’s arteritis is a chronic inflammatory condition that affects the largest blood vessel in the body (the aorta) and its branches. Thus, the complications of Takayasu’s arise directly or indirectly from damage to these blood vessels. The vasculitides are classified according to the size of blood vessel involved. Takayasu’s is the classic “large vessel” vasculitis.
Pictured below is a normal aortic arch on the left, with narrow, smooth blood vessels. On the right is an example of an abnormal aortic arch in a patient with Takayasu’s, with obvious dilation of the ascending aorta on the left side of the picture.

Clinicians divide Takayasu’s arteritis into two phases: 1) a systemic phase; and 2) an occlusive phase. Although these two phases are not always distinct (i.e., patients may have features of both phases at the same time), this division is a useful way of thinking about the disease.
In the systemic phase, patients have symptoms and signs of an active inflammatory illness. These may include “constitutional symptoms” (fever, fatigue, weight loss), arthritis, and non-specific aches and pains. There may be tenderness overlying affected arteries. Most patients have elevations of the erythrocyte sedimentation rate during the systemic phase.
The systemic phase is succeeded by the occlusive phase, during which patients begin to develop symptoms caused by the narrowing of affected arteries. These may include pain in the limbs that occurs during repetitive activities (“claudication”), such as pain in the arm that occurs while using a handsaw or pain in the calves brought on by walking. The symptoms also include dizziness upon standing up, headaches, and visual problems. During the occlusive phase, affected blood vessels may be narrowed to such an extent that the normal arterial pulsations (“pulses”) in the neck, elbow, wrist, or lower extremities cannot be felt. Using a stethoscope, one may also hear “bruits” (pronounced ‘brew eez’), a harsh, “whooshing” sounds made by the flow of blood through abnormally narrowed vessels. High blood pressure is common, but blood pressures taken in the arms may be read as falsely low if there is a narrowing of an artery high up in the arm. With some patients, it is not possible to get accurate blood pressure readings in the arms. Using an ophthalmoscope, a physician may observe characteristic malformations of blood vessels that occur in advanced cases of Takayasu’s arteritis.
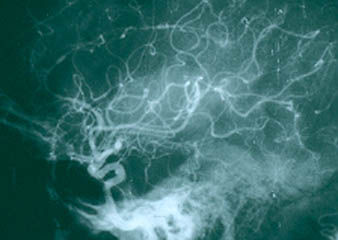
Although the lung involvement in Takayasu’s is frequently overshadowed by involvement of systemic large blood vessels, the pulmonary arteries may also be affected in this disorder. Pictured below is a pulmonary angiogram demonstrating beading and cut–off lesions of the right pulmonary arteries, and a large aneurysm of the left pulmonary artery.

What Causes Takayasu’s Arteritis?
The cause of Takayasu’s arteritis is not known. Some evidence suggests that an infection of some kind — viral, bacterial, or other — occurring in a person with other predisposing factors (such as the correct genes), may lead to this disease. This is an attractive hypothesis, but definitive evidence for it is lacking.
How is Takayasu’s Arteritis Diagnosed?
Making the diagnosis of Takayasu’s arteritis can be extremely difficult. Unfortunately it is very common for the disease to smoulder in the walls of large blood vessels for years, causing only non-specific symptoms associated with the systemic phase of the illness (or no symptoms), until a major complication results. These major complications may include dilation of the aorta with “stretching” of the aortic valve in the heart; critically reduced blood flow to an arm or leg; a stroke caused by high blood pressure in vessels of the brain, and many others.
Once the diagnosis is suspected, it is usually confirmed by a radiographic procedure such as an angiogram or a magnetic resonance imaging study demonstrating significant large artery disease consistent with Takayasu’s. In some cases in which blood vessel damage is so severe as to necessitate surgery to repair the aortic valve, the aorta, or some other large blood vessel, physicians are able to make unequivocal diagnoses by looking at tissue from the involved blood vessels under the microscope. Takayasu’s arteritis is pathologically indistinguishable from giant cell arteritis. In both, destruction of the blood vessel wall and giant cells are frequently present.
Pictured below is an example of large artery involvement in Takayasu’s arteritis. Magnetic resonance imaging study of the aorta and large blood vessels of the upper extremities, showing a large aneurysm of the ascending aorta, blockage of the right axillary artery (note the interruption of blood flow near the shoulder on the left of the figure), and many narrowings of the left subclavian artery (on the right of the figure).

Treatment and Course of Takayasu’s Arteritis
The great majority of patients with Takayasu’s arteritis respond to prednisone. The usual starting dose is approximately 1 milligram per kilogram of body weight per day (for most people, this is approximately 60 milligrams a day). Because of the significant side–effects of long-term high–dose prednisone use, the starting dose is tapered over several weeks to a dose that the physician feels is tolerable for the patient.
For long–term treatment in addition to prednisone (as “steroid sparing agents”), methotrexate, azathioprine, and even cyclophosphamide are sometimes used. There have been few studies of the use of these medications in this disease.
What’s New in Takayasu’s Arteritis?
One of the biggest problems confronting Takayasu’s patients and the physicians who care for them is determining how active the disease is. This can be an exceptionally challenging problem. The erythrocyte sedimentation rate (ESR) probably remains the most reliable marker of disease activity, but even this test is not helpful in a sizeable number of patients who have active arterial inflammation but normal ESRs. Because the treatments for Takayasu’s arteritis may be associated with substantial side–effects, we need more accurate means of gauging disease activity.
To this end, a study conducted by the International Network for the Study of Systemic Vasculitides (“INSSYS”) may be helpful. Investigators from INSSYS, which includes more than 300 physicians, scientists, and other experts in vasculitis from more than 50 different medical centers across the world, have been conducting a “Surrogate Markers Study” for the past several years. In this study, the investigators examine blood specimens from patients with vasculitis for the purpose of identifying proteins and other molecules whose presence indicates ongoing inflammation. Improved understanding of these diseases at a molecular level may permit more rational use of treatments in the future.
In medical terms, by David Hellmann, M.D.
A discussion of Takayasu’s Arteritis written in medical terms by David Hellmannn, M.D. (F.A.C.P.), The Johns Hopkins Vasculitis Center, for the Rheumatology Section of the Medical Knowledge Self-Assessment Program published and copyrighted by the American College of Physicians (Edition 11, 1998). The American College of Physicians has given us permission to make this information available to patients contacting our Website.
Takayasu’s arteritis is a granulomatous vasculitis chiefly of young women that involves the aorta and its major branches. Patients can present initially with obscure systemic symptoms such as fever of unknown origin or more commonly with symptoms and signs of large vessel vasculitis such as hypertension from renal artery stenosis, aortic regurgitation from aortitis, or stroke from carotid artery occlusion. Bruits and diminished or absent pulses are the most reliable signs. Anemia and elevated ESR accompany active disease. Diagnosis is confirmed by angiography showing stenosis and dilation of the aorta, its branches, or both. Thickening of the aortic wall detectable by MRI or ultrasonography can precede angiographic changes. Prednisone is effective for the systemic symptoms and can thwart progression of the vasculitis. Methotrexate may be an effective corticosteroid-sparing agent. Angioplasty alleviates renal artery stenosis about half the time. When needed, vascular bypass procedures and aortic valve replacement usually work well if deferred until the disease is inactive. Estimating disease activity is difficult but is based on systemic symptoms, anemia, ESR, progression of lesions, and pathology (when available).